O que são esclerodermia e esclerose sistêmica?
A esclerodermia, cujo nome é derivado das palavras gregas skleros (endurecido) e derma (pele), é uma doença de pele de origem autoimune, caracterizada, como o próprio nome sugere, por um progressivo enrijecimento da pele.
Quando além do endurecimento da pele, o paciente também apresenta envolvimento de vasos sanguíneos e órgãos internos, chamamos a doença de esclerose sistêmica.
A esclerodermia é um conjunto de doenças que apresentam manifestações clínicas distintas, mas que têm como característica comum o enrijecimento da pele. A doença é tradicionalmente classificada com base na extensão do envolvimento da pele e no padrão de envolvimento de órgãos internos. Conforme veremos a seguir, os diferentes subtipos de esclerose sistêmica estão associados a diferentes padrões de envolvimento de órgãos e de gravidade da doença.
A esclerodermia é uma doença relativamente rara, que acomete cerca de 200 a 300 pessoas a cada 1 milhão de habitantes. A doença é 4 vezes mais frequente nas mulheres do que nos homens e geralmente surge entre os 25 e os 50 anos.
Classificação da esclerodermia
Há dois tipos principais de esclerodermia:
- Esclerodermia localizada (forma mais comum nas crianças).
- Esclerodermia sistêmica, também chamada esclerose sistêmica (forma mais comum nos adultos).
Esclerodermia localizada
A esclerodermia localizada é uma doença que se restringe à pele, podendo em alguns casos se estender até músculos, articulações e ossos. A esclerodermia localizada não apresenta envolvimento de órgão internos nem lesões vasculares graves.
A esclerodermia localizada é subdividida em dois tipos:
Esclerodermia linear
A esclerodermia linear, forma mais comum em crianças e adolescentes, caracteriza-se por lesões de fibrose, com espessamento e endurecimentos da pele em bandas ou linhas, localizadas no tronco ou membros, que costumam ficar restritas a apenas uma metade do corpo.

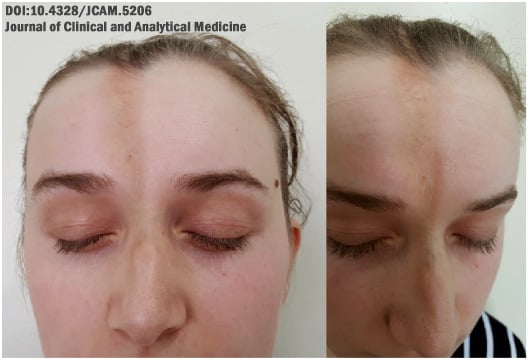
Uma forma típica de esclerodermia linear é a chamada lesão em “golpe de sabre” ou “coup de sabre”, que é um espessamento da pele que costuma surgir na região do crânio e parece ter sido feito por um golpe de espada.
A lesão pode ser hipopigmentada (mais clara que a pele) ou hiperpigmentada (mais escura que a pele).
Morfeia
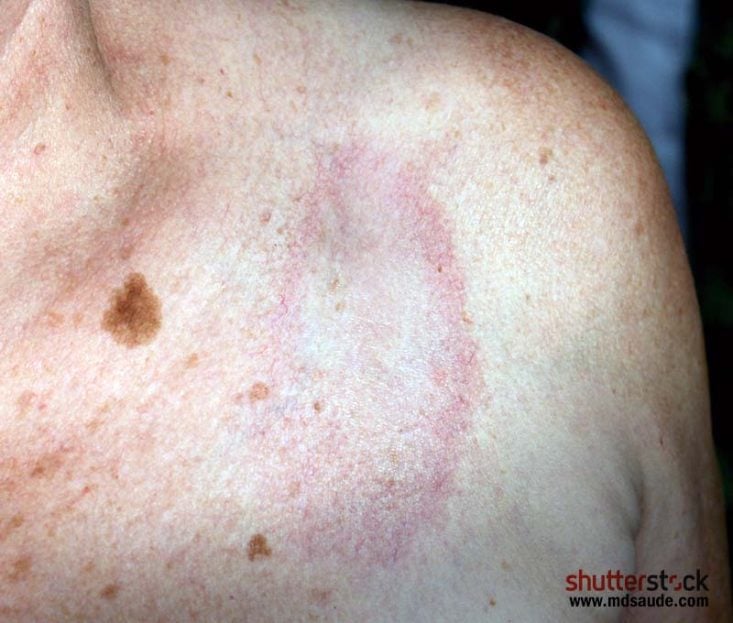
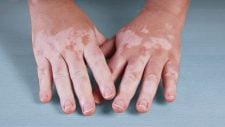
Morfeia é habitualmente a forma clínica mais branda de esclerodermia e se apresenta como zonas de pele espessada, geralmente de forma oval, com centro mais esbranquiçado e bordas violáceas. Com o tempo, as lesões podem se tornar mais acastanhadas. Estas lesões surgem com mais frequência no tronco e nos membros.
A morfeia generalizada é um subtipo mais grave, com múltiplas placas espessadas que acometem extensas áreas da pele, que podem ter uma coloração mais esbranquiçada ou amarelada, sendo habitualmente mais claras que as áreas de pele saudável.
Esclerose sistêmica
A esclerose sistêmica é a forma mais grave de esclerodermia, havendo não só acometimento da pele, mas também de vasos sanguíneos e órgãos internos. A esclerose sistêmica pode ser subdividida em três formas:
Esclerose sistêmica cutânea difusa
Na esclerose sistêmica cutânea difusa, além do acometimento da pele, que costuma ser difuso, porém restrito às regiões centrais do corpo, como tronco e início dos membros, essa forma de esclerodermia também pode afetar vários órgãos internos, tais como pulmões, coração, sistema gastrointestinal e rins. Essa forma de esclerodermia é bem grave e, se não for tratada, costuma ser fatal.
Esclerose sistêmica cutânea limitada
Na esclerose sistêmica cutânea limitada, o acometimento da pele costuma ser mais periférico, ficando restrito a mãos, antebraços, pernas, face e pescoço. Partes centrais, como tronco, braços e coxas, costumam ser poupados.
O atingimento dos órgãos internos costuma ser mais tardio e tende a ficar confinado, quase sempre, ao tubo digestivo e aos pulmões.
A esclerose sistêmica cutânea limitada costuma causar a chamada síndrome CREST, que é um acrônimo em inglês para as seguintes complicações:
- Calcinose: depósitos de cálcio na pele;
- fenômeno de Raynaud: alterações vasculares que provocam isquemia e mudanças temporárias na cor dos dedos.
- Esôfago: alterações na motilidade do esôfago.
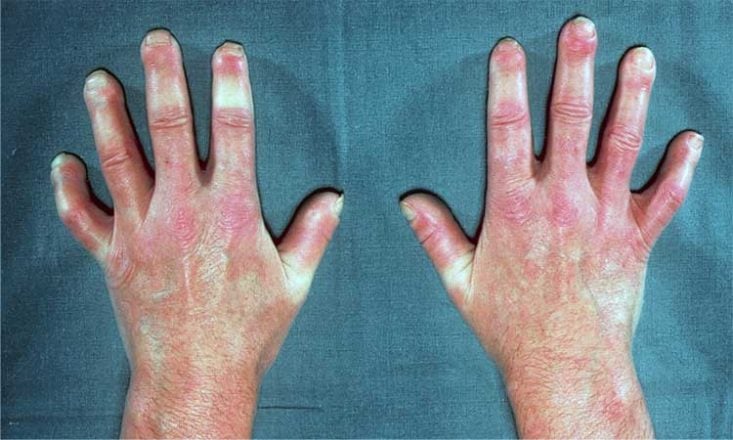
- eSclerodactilia (Sclerodactyly em inglês): espessamento e enrijecimento da pele dos dedos.
- Telangiectasias: vasos capilares finos, vermelhos ou violáceos que se aglomeram na superfície da pele.
Esclerose sistêmica sine esclerodermia
A esclerose sistêmica sine esclerodermia é uma forma rara, na qual o paciente tem o quadro de esclerose sistêmica, mas sem o acometimento da pele.
Causas
A esclerodermia é uma doença de origem autoimune, ou seja, é uma doença na qual o nosso sistema imunológico inapropriadamente ataca as nossas próprias células. O mecanismo exato de como a doença se desenvolve é bem complexo e ainda não está totalmente esclarecido. Sabemos, porém, que a esclerose sistêmica é o resultado de três processos patológicos distintos:
- Lesões inflamatórias das pequenas artérias e arteríolas.
- Anormalidades na resposta imunológica frente a essas lesões vasculares.
- Produção excessiva de colágeno, que se deposita na pele e nos órgãos internos, levando ao seu espessamento e enrijecimento.
Não sabemos ainda o motivo pelo qual a esclerodermia surge, mas a suscetibilidade genética parece desempenhar um papel relevante. O problema é que a genética sozinha não é suficiente para a doença surgir.
A teoria atualmente mais aceita é a de que pessoas geneticamente susceptíveis, ou seja, pessoas que carregam certos genes, quando são expostos a determinados fatores ambientais, sejam eles infecções ou substâncias químicas, ativam o gatilho que desencadeia os processos patológicos que levam à esclerose sistêmica.
Algumas bactérias e vírus podem apresentar proteínas que são semelhantes àquelas presentes no nosso corpo. Em pessoas susceptíveis, o sistema imunológico pode tentar criar anticorpos contra essas proteínas virais ou bacterianas, mas, de forma equivocada, acaba por criar anticorpos contra as nossas próprias proteínas. Infecções por citomegalovírus, parvovírus B19 e herpes vírus 5 são alguns dos gatilhos sugeridos.
O mesmo processo pode ser desencadeado pela exposição ocupacional a certas substâncias químicas, como a sílica ou produtos derivados do petróleo (cloreto de vinila, tricloroetileno, tolueno, resinas, benzeno ou tetracloreto de carbono). Também há relatos de desenvolvimento de esclerodermia em pacientes submetidos à quimioterapia com bleomicina.
Sintomas
Os sintomas da esclerodermia dependem da forma de doença que o paciente tem.
Esclerodermia localizada
Na esclerodermia localizada, os sintomas praticamente se restringem à pele. A maioria dos pacientes com morfeia desenvolve apenas uma ou duas lesões, que consistem em espessamento, enrijecimento e mudanças de cor da pele.
Essas alterações podem durar anos, mas tendem a melhorar. Essa forma de esclerodermia é branda e o prognóstico a longo prazo é bom, pois a doença torna-se inativa, ficando o paciente curado.
Na morfeia generalizada, o número de lesões de pele é maior e elas tendem a se confluir, acometendo grandes áreas do corpo. Lesões que causam desfiguração podem ocorrer, e mesmo quando há melhora espontânea após alguns anos, as manchas tendem a ser persistentes.
Como já mencionado, a esclerodermia linear é mais comum em crianças e adolescentes. Cerca de 80% dos pacientes têm menos de 20 anos. A cada 5 pessoas acometidas, 4 são mulheres.
A esclerodermia linear tem um potencial de complicações maior que a morfeia. O espessamento da pele pode ser profundo, o que em crianças pode comprometer o crescimento de ossos e articulações. A esclerodermia linear costuma permanecer ativa por dois a cinco anos, mas em alguns pacientes ela pode durar mais tempo. A esclerodermia em golpe de espada é a forma com maior potencial de provocar lesões desfigurantes, pois ela afeta preferencialmente a face e o crânio.
Ao contrário da morféia, a esclerodermia linear pode recorrer mesmo após um longo período de inatividade.
Esclerose sistêmica
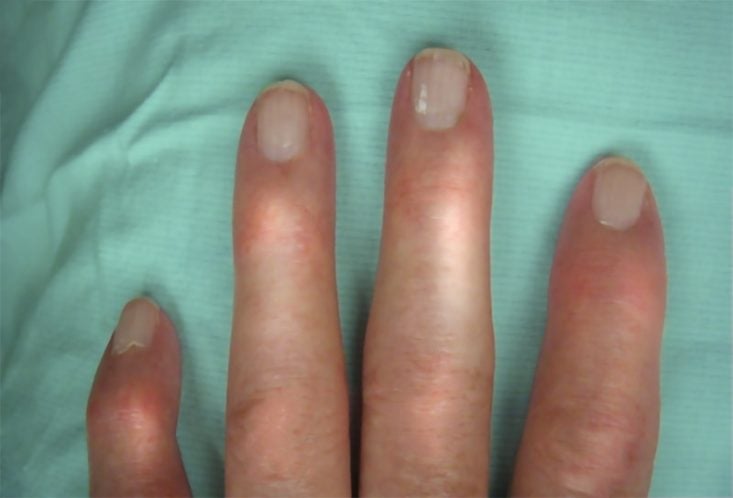
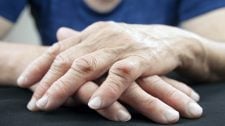
Também na esclerose sistêmica os sintomas cutâneos são os mais comuns. Entre eles, um que está presente em quase todos os casos é o chamado fenômeno de Raynaud.
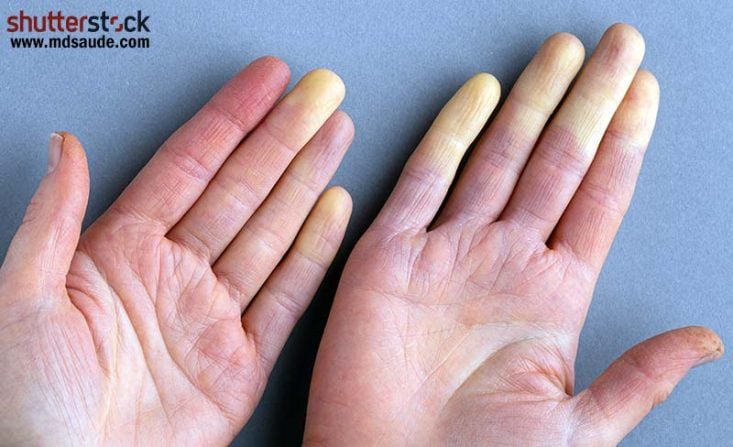
O fenômeno de Raynaud é distúrbio vascular, definido como alterações sequenciais de cor que ocorrem geralmente nos dedos das mãos, precipitados por frio, estresse ou simplesmente mudanças de temperatura ambiente. As alterações de cor seguem a seguinte ordem:
- Palidez: a mão fica mais branca por uma súbita vasoconstrição arterial, que diminui o aporte de sangue.
- Acrocianose: a mão fica azulada ou arroxeada, conforme a falta de sangue vai se prolongando.
- Hiperemia de reperfusão: a mão fica avermelhada e quente devido à súbita vasodilatação e restabelecimento da circulação normal de sangue.
O fenômeno de Raynaud pode surgir vários anos antes do início dos sintomas da esclerose sistêmica cutânea limitada. Já na esclerose sistêmica cutânea difusa, o seu surgimento costuma ocorrer com o restante do quadro clínico. Em alguns casos, a fase de isquemia pode ser grave e levar à necrose das pontas dos dedos.
É importante destacar que o fenômeno de Raynaud também pode ocorrer em pessoas que não tenham esclerodermia. Ele de forma alguma é um sinal exclusivo da esclerodermia.
Falamos mais sobre o fenômeno de Raynaud no artigo: Fenômeno de Raynaud: o que é, causas, sintomas e tratamento.
Além do Raynaud, outras alterações de pele da esclerose sistêmica são:
- Espessamento e enrijecimento da pele, criando um aspecto de pele lisa, brilhante e dura.
- Coceira: mais comum nos estágios iniciais da doença.
- Inchaços: também mais comum nos estágios iniciais da doença.
- Hiperpigmentação ou despigmentação da pele (pele em “sal e pimenta”).
- Perda de cabelo.
- Esclerodactilia.
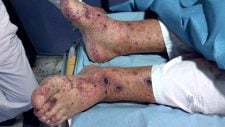
- Úlceras nos dedos.
- Dor na ponta dos dedos.
- Telangiectasias.
- Calcinose cutânea.
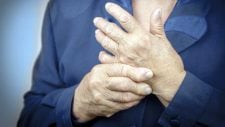
Além da lesão de pele, que é praticamente universal a todas as formas de esclerodermia, o paciente com esclerose sistêmica também costuma apresentar uma gama de outros sintomas. Entre os mais comuns, podemos citar:
- Fadiga (76%).
- Rigidez das articulações (74%).
- Perda de força (68%).
- Dor (67%).
- Dificuldade para dormir (66%).
Em relação ao acometimento dos órgãos, os mais comuns são:
- Alterações da motilidade do esôfago, com dificuldade para engolir.
- Esofagite.
- Refluxo gastroesofágico.
- Diarreia.
- Prisão de ventre.
- Incontinência fecal.
- Hemorragia digestiva.
- Doença intersticial pulmonar.
- Hipertensão pulmonar.
- Câncer de pulmão.
- Insuficiência renal aguda.
- Hipertensão maligna.
- Arritmias cardíacas.
Tratamento
Não existe tratamento que leve à cura da esclerodermia. Como já referido, as formas limitadas à pele podem ter regressão espontânea após alguns anos.
Já a esclerose sistêmica é uma doença de evolução lenta, mas permanente. O seu tratamento visa aliviar os sintomas e reduzir a atividade da doença.
Os fármacos utilizados dependem dos sintomas que o paciente tem. Exemplos:
- Vasodilatadores, como a nifedipina, costumam ser usados para tratar o fenômeno de Raynaud.
- Inibidores da bomba de prótons, como o omeprazol, ajudam nos sintomas gástricos e do refluxo.
- Anti-inflamatórios e corticoides em doses baixas ajudam na dor e na rigidez articular.
- Imunossupressores, como metotrexato ou micofenolato mofetil, ajudam a reduzir os danos da pele e órgãos causados pelo sistema imunológico.
- Anti-hipertensivos para tratar a hipertensão.
Não existe nenhum tratamento que seja universal para todas as formas de esclerodermia. Cada tratamento deve ser instituído individualmente, conforme os sintomas e o tipo de envolvimento de órgão existente em cada paciente.
Referências
- 2013 classification criteria for systemic sclerosis: an American college of rheumatology / European league against rheumatism collaborative initiative – Annals of the Rheumatic Diseases.
- Update of EULAR recommendations for the treatment of systemic sclerosis – Annals of the Rheumatic Diseases.
- BSR and BHPR guideline for the treatment of systemic sclerosis – Rheumatology.
- Clinical manifestations and diagnosis of systemic sclerosis (scleroderma) in adults – UpToDate.
- Overview of the treatment and prognosis of systemic sclerosis (scleroderma) in adults – UpToDate.
- Pathogenesis of systemic sclerosis (scleroderma) – UpToDate.
- What is scleroderma? – Scleroderma Foundation.
- The case of frontal linear scleroderma (en coup de sabre) – Journal of Clinical and Analytical Medicine.

Dúvidas de leitores sobre este tema
Perguntas enviadas por leitores e selecionadas pelo editor por sua relevância para este artigo.
Mais comentários dos leitores
Eu Silvana, descobri esclerodermia sistemica há 4 meses, após uma longa batalha de dois anos de diarréia, cheguei aos 35 kls, com 1.72, fui internada, 18 dias na UTI para descobrirem o problema.
Quando descobri fiquei muito mal, eu parecia aneurexa, magreza extrema, porque a esclero pode dar desnutrição.
HOje me trato com um reumato Dr. Jean maravilhoso, estou medicada, doença controlada.
Estou com 60 kls, meu corpo voltou, está lisinho e bonito.
Eu foco no lado bom da doença, pela boa, sem ruga, sem flacidez.
Gracas a Deus meus orgão internos estáo 100%.
bom dia DR. meu filho tem 13 anos e esta fazendo uma bateria de exames pois ainda nao sabemos o tipo de esclerodermia , mas ja tem o diagnostico, aparentimente remete nos a localizada, sucede que os medicos ainda nao deram nehuma medicacao e eu so tenho lhe aplicado cremes para hidratar a pele , peco ajuda com a indicacao de cremes, ele costuma sentir uma comichao interna
Apareceu uma mancha marron em minha barriga, fiz uma biópsia e o resultado foi fibroesclerose dérmica a médica não passou nada para eu tomar, estou sentindo as pontas dos dedos com dormência isso faz parte da doença, que médico devo procurar para acompanhamento
Chamo-me Carla d’Oliveira. Sou portadora de esclerodermia do tipo Morfeia desde os 9 anos, tendo sido diagnosticado em Portugal (Faro) aos 16 anos. Tenho 44 anos e tenho alguns dos sintomas que refere, nomeadamente insónia. No entanto nos últimos meses sinto dor na pele onde tenho a lesão (fossa elíaca esquerdo), é comum?
Obrigada por organizar as informações de maneira a facilitar a leitura e o conhecimento. Tenho ES, mais especificamente CREST, que está até bem controlada, mas sempre busco ler e aprender mais, mesmo tendo acompanhamento de reumatologista. Obrigada mesmo.
Que médico eu devo procurar para tratar da morfeia
Dr. Bom dia fui diagnosticado com 15 anos com esclerodermia em golpe de sabre e na quela época me tratava no hospital do fundão hoje estou com 34 anos e a doença está inativa . Contudo tenho um filho de 2 anos e tenho muito medo dele venha a passar por tudo que passei , teria algum exame específico para avaliar se meu filho vai ter essa mesma doença? Algo para previnir dessa doença? Muito obrigado
Att Tales Lima
Eu moro em Blumenau descobri a esclerodermia a quase um ano, tenho a cutânea e a sistêmica, estou com vários problemas gastrite, esofagite, faringite, laringite, diarreia, tosse seca constante e incontinência urinária não consigo dormir mais que 3 horas meu cabelo caiu muito na frente e a cabeça coça e arde muito aqui os médico não sabem muito a respeito e ando com todos os exames sempre querem tratar como Alergia ou gripe. Não sei muito o que fazer quando tenho essas crises de tosse, incontinência urinária e diarreia. Alguma sugestão de como me livrar da tosse?
Minha amiga faleceu de esclerodermia há 4 anos atrás, com 34 anos, foi uma luta muito dura e triste, ela deixou duas crianças lindas, um menino que na época tinha 1 ano e a menina 4 anos. Essa doença é terrivel .
Olá. Eu tenho morféia, esclerodermia em placas. Causada por infecção por borrelias burgdorferi, ou borrelia brasileira, transmitida por picada de carrapato infectado. Sonho com o dia em que todos os doentes sejam submetidos ao exame Sorologia para borreliose, Western Blot, para verificar a hipótese de Doença de Lyme, à qual fui acometida há 12 anos e faço tratamento há 10 anos. Além de mim há outros infectados por borrelias que desenvolveram todos os tipos de esclerodermia.
Meu irmão tem essa doença e está atingindo os órgãos internos, o que pode fazer para melhorar a situação dele?
Descobri há um ano esclerodermia em.minha filha de 6anos; no início pensava ser uma mancha simples vermelha de um esbarrao qualquer; daí foi ficando marrom invés de arroxear; depois preta; depois esfumacada;levei em vários médicos; uns falaram ser queimaduras de sumo de frutas; outro trombose; daí uma dermatologista descobriu por meio da biopsia que se tratava de esclerodermia localizada; eu quase enlouqueci ao pesquisar na net sobre a doença; mais a médica não me esclareceu nada sobre a doença; parecia ser algo novo p ela tb; minha menina tá com 7 anos ; toma tecnomet; 3 dias na semana; queria saber mais sobre o que fazer; e até quantos anos corre o risco de sair mais lesões em seu corpo?
E o que posso fazer?
Por favor Dr. Me ajude e desde já obrigada
Corticoides são uma boa opção de tratamento para a esclerodermia?