¿Qué son la esclerodermia y la esclerosis sistémica?
La esclerodermia, cuyo nombre deriva del griego (sklēródermos «piel endurecida»), es una enfermedad de la piel de origen autoinmune que se caracteriza, como su nombre indica, por un progresivo endurecimiento de la piel.
Cuando además del endurecimiento de la piel, el paciente también presenta afectación de vasos sanguíneos y órganos internos, denominamos a la enfermedad esclerosis sistémica.
La esclerodermia es un conjunto de enfermedades que presentan manifestaciones clínicas distintas, pero que tienen como característica común el endurecimiento de la piel. La enfermedad se clasifica tradicionalmente según la extensión de la afectación de la piel y el patrón de afectación de los órganos internos. Como veremos a continuación, los diferentes subtipos de esclerosis sistémica se asocian con diferentes patrones de afectación de órganos y gravedad de la enfermedad.
La esclerodermia es una enfermedad relativamente rara, que afecta a unas 200 a 300 personas por millón de habitantes. La enfermedad es 4 veces más frecuente en mujeres que en hombres y suele aparecer entre los 25 y los 50 años.
Clasificación de la esclerodermia
Hay dos tipos principales de esclerodermia:
- Esclerodermia localizada (forma más común en niños).
- Esclerodermia sistémica, también llamada esclerosis sistémica (forma más frecuente en adultos).
Esclerodermia localizada
La esclerodermia localizada es una enfermedad que se limita a la piel y en algunos casos puede extenderse a los músculos, articulaciones y huesos. La esclerodermia localizada no tiene implicación con órganos internos ni lesiones vasculares graves.
La esclerodermia localizada se subdivide en:
1. Esclerodermia lineal
La esclerodermia lineal, la forma más común en niños y adolescentes, se caracteriza por lesiones de fibrosis, con engrosamiento y endurecimiento de la piel en bandas o líneas, localizadas en el tronco o las extremidades, que suelen estar restringidas a solo la mitad del cuerpo.

Una forma típica de esclerodermia lineal es la lesión denominada “golpe de sable” o “coup de sabre”, que es un engrosamiento de la piel que suele aparecer en la región del cráneo y parece haber sido realizado por un golpe de espada.
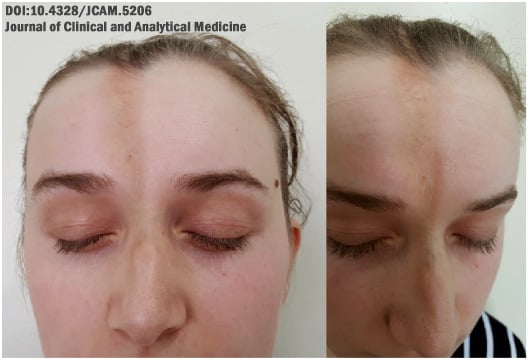
La lesión puede ser hipopigmentada (más clara que la piel) o hiperpigmentada (más oscura que la piel).
2. Morfea
La morfea suele ser la forma clínica más leve de esclerodermia y se presenta como áreas de piel engrosada, generalmente de forma ovalada, con un centro blanquecino y bordes violáceos. Con el tiempo, las lesiones pueden volverse más marrones. Estas lesiones aparecen con mayor frecuencia en el tronco y las extremidades.
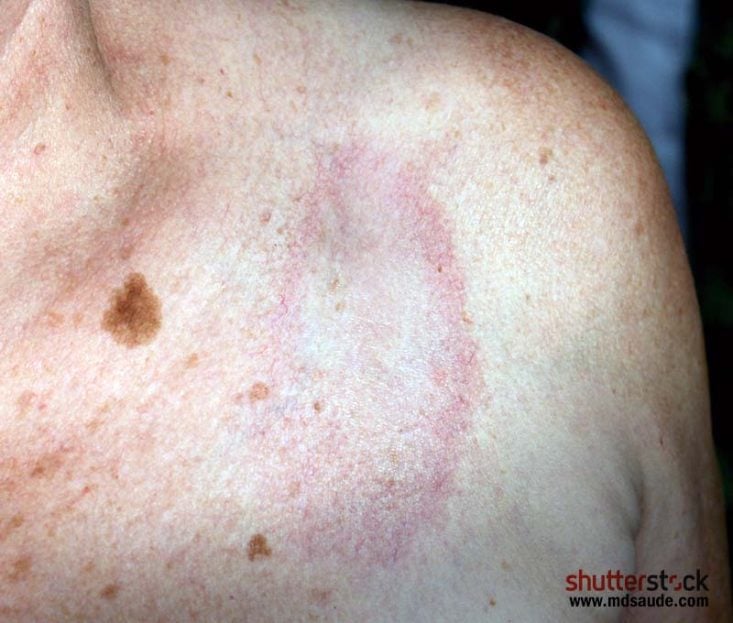
La morfea generalizada es un subtipo más grave, con múltiples placas engrosadas que afectan a grandes zonas de la piel, que pueden ser de color más blanquecino o amarillento, y suelen ser más claras que las zonas de piel sana.
Esclerosis sistémica
La esclerosis sistémica es la forma más severa de la enfermedad, que no solo acomete la piel, sino también los vasos sanguíneos y órganos internos. La esclerosis sistémica puede subdividirse en tres formas:
1. Esclerosis sistémica cutánea difusa
La esclerosis sistémica cutánea difusa, además de la afectación de la piel (que suele ser difusa, pero restringida a regiones centrales del cuerpo, como el tronco y comienzo de las extremidades) también puede afectar a varios órganos internos, como los pulmones, corazón, sistema gastrointestinal y riñones. Esta forma de esclerodermia es muy grave y, si no se trata, suele ser mortal.
2. Esclerosis sistémica cutánea limitada
En la esclerosis sistémica cutánea limitada, el acometimiento de la piel suele ser más periférico, quedando restricto a las manos, antebrazos, piernas, vertiente y cuello. Partes céntricas, como tronco, brazos y muslos, generalmente son ahorrados.
El acometimiento de los órganos internos suele ser más tardío y tiende a limitarse, casi siempre, al tubo digestivo y a los pulmones.
La esclerosis sistémica cutánea limitada generalmente causa el llamado síndrome CREST, que es un acrónimo en inglés para las siguientes complicaciones:
- Calcinosis: depósitos de calcio en la piel;
- fenómeno de Raynaud: alteraciones vasculares que provocan isquemia y cambios temporales en el color de los dedos;
- Esófago: alteraciones en la motilidad del esófago;
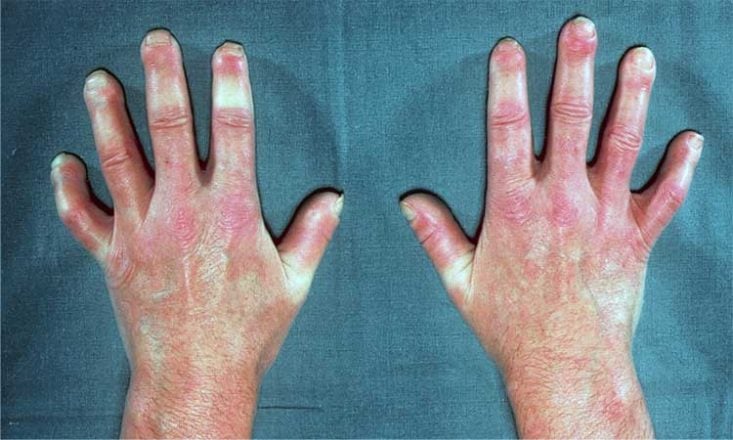
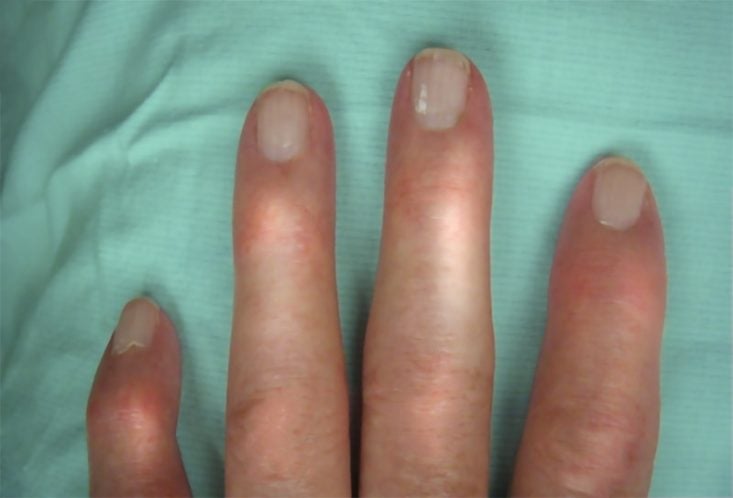
- eSclerodactilia (Sclerodactyly en inglés): engrosamiento y rigidez de la piel de los dedos;
- Telangiectasias: vasos capilares delgados, rojos o violáceos que se aglomeran en la superficie de la piel.
3. Esclerosis sistémica sine esclerodermia
La esclerosis sistémica sine esclerodermia es una forma rara, en la que el paciente tiene esclerosis sistémica, pero sin afectación de la piel.
Causas
La esclerodermia es una enfermedad de origen autoinmune, es decir, es una enfermedad en la que nuestro sistema inmunitario ataca de manera inapropiada a nuestras propias células. El mecanismo exacto de cómo se desarrolla la enfermedad es bastante complejo y aún no se comprende completamente. Sin embargo, sabemos que la esclerosis sistémica es el resultado de tres procesos patológicos distintos:
- Lesiones inflamatorias de las pequeñas arterias y arteriolas.
- Anomalías en la respuesta inmune frente a estas lesiones vasculares.
- Producción excesiva de colágeno, que se deposita en la piel y órganos internos, llevando a su engrosamiento y rigidez.
No sabemos todavía el motivo por lo que aparece la esclerodermia, pero la susceptibilidad genética parece jugar un papel importante. El problema es que solamente la genética no es suficiente para que la enfermedad se presente.
La teoría actualmente más aceptada es que las personas genéticamente susceptibles, es decir, personas que tienen determinados genes, cuando se exponen a ciertos factores ambientales, ya sean sustancias químicas o infecciones, activan el gatillo que dispara los procesos patológicos que llevan a la esclerodermia sistémica.
Algunas bacterias y virus pueden presentar proteínas que son similares a las presentes en nuestro cuerpo. En personas susceptibles, el sistema inmune puede tratar de crear anticuerpos contra estas proteínas virales o bacterianas, pero por error termina creando anticuerpos contra nuestras propias proteínas. Infecciones por citomegalovirus, parvovirus B19 y herpes virus 5 son algunos de los disparadores sugeridos.
El mismo proceso puede ser desencadenado por la exposición ocupacional a ciertas sustancias químicas, tales como sílice o productos derivados del petróleo (cloruro de vinilo, tricloroetileno, tolueno, resinas, benceno o tetracloruro de carbono). Hay también informes del desarrollo de esclerodermia en pacientes sometidos a quimioterapia con bleomicina.
Síntomas
Los síntomas de la esclerodermia dependen de la forma de enfermedad que el paciente tiene.
Esclerodermia localizada
En la esclerodermia localizada, los síntomas prácticamente se restringen a la piel. La mayoría de los pacientes con morfea desarrolla solamente una o dos lesiones, que consisten en engrosamiento, rigidez y cambio de color de la piel.
Estos cambios pueden durar años, pero tienden a mejorar. Esta forma de esclerodermia es leve y el pronóstico a largo plazo es bueno, ya que la enfermedad se vuelve inactiva y el paciente se cura.
En la morfea generalizada, el número de lesiones de piel es mayor y ellas tienden a fusionarse, afectando a grandes áreas del cuerpo. Pueden ocurrir lesiones que desfiguran, e incluso cuando hay una mejoría espontánea después de algunos años, las manchas tienden a ser persistentes.
Como ya se mencionó, la esclerodermia lineal es más común en niños y adolescentes. Alrededor del 80% de los pacientes tienen menos de 20 años. De cada 5 personas afectadas, 4 son mujeres.
La esclerodermia lineal tiene un potencial de complicaciones mayor que la morfea. El engrosamiento de la piel puede ser profundo, lo que en los niños puede afectar al crecimiento de los huesos y las articulaciones. La esclerodermia lineal generalmente permanece activa durante dos a cinco años, pero en algunos pacientes puede durar más tiempo.
La esclerodermia en golpe de sable es la forma que tiene el mayor potencial para causar lesiones deformantes, ya que afecta a la cara y al cráneo.
A diferencia de la morfea, la esclerodermia lineal puede volver inclusive después de un largo período de inactividad.
Esclerosis sistémica
También en la esclerosis sistémica, los síntomas cutáneos son los más comunes. Entre ellos, uno que está presente en casi todos los casos se llama fenómeno de Raynaud.
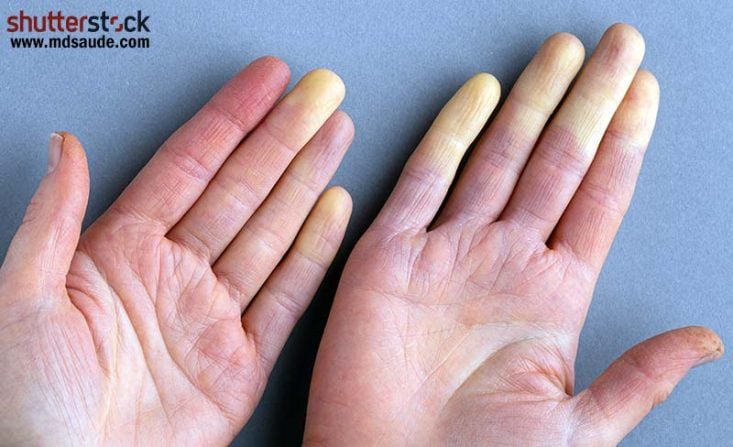
El fenómeno de Raynaud es un trastorno vascular, definido como cambios de color secuenciales que se producen en los dedos, generalmente precipitados por el frío, estrés o simplemente cambios en la temperatura ambiente. Los cambios de color siguen el siguiente orden:
- Palidez: la mano se queda blanca por una repentina vasoconstricción arterial, que disminuye la cantidad de sangre.
- Acrocianosis: la mano se queda azulada o violácea, a medida que la falta de sangre va prolongándose.
- Hiperemia de reperfusión: la mano se enrojece y se calienta debido a la vasodilatación repentina y al restablecimiento de la circulación sanguínea normal.
El fenómeno de Raynaud puede presentarse varios años antes del inicio de los síntomas de la esclerosis sistémica cutánea limitada. Ya en la esclerosis sistémica cutánea difusa, su aparición ocurre generalmente junto con el resto del cuadro clínico. En algunos casos, la fase de isquemia puede ser grave y llevar a la necrosis de las yemas de los dedos.
Es importante señalar que el fenómeno de Raynaud también puede ocurrir en personas que no tienen esclerodermia. De ninguna manera es un signo exclusivo de esclerodermia.
Además del Raynaud, otros cambios de la piel en la esclerosis sistémica son:
- Engrosamiento y endurecimiento de la piel, creando un aspecto de piel lisa, brillante y dura.
- Picazón: más común en las primeras etapas de la enfermedad.
- Hinchazón: también más común en las primeras etapas de la enfermedad.
- Hiperpigmentación o despigmentación de la piel (piel en “sal y pimienta”).
- Pérdida del cabello.
- Esclerodactilia.
- Úlceras en los dedos.
- Dolor en la punta de los dedos.
- Telangiectasias.
- Calcinosis cutánea.
Además de la lesión de la piel, que es prácticamente universal para todos los tipos de esclerodermia, el paciente con esclerosis sistémica también presenta una gama de otros síntomas. Entre los más comunes, podemos mencionar:
- Fatiga (76%).
- Rigidez de las articulaciones (74%).
- Pérdida de fuerza (68%).
- Dolor (67%).
- Dificultad para dormir (66%).
Con respecto al acometimiento de los órganos, los más comunes son:
- Cambios de la motilidad del esófago, con dificultad para tragar.
- Esofagitis.
- Reflujo gastroesofágico.
- Diarrea.
- Estreñimiento.
- Incontinencia fecal.
- Hemorragia digestiva.
- Enfermedad intersticial pulmonar.
- Hipertensión pulmonar.
- Cáncer de pulmón.
- Insuficiencia renal aguda.
- Hipertensión maligna.
- Arritmias cardíacas.
Tratamiento
No hay tratamiento que lleve a la cura de la esclerodermia. Como ya se mencionó, las formas limitadas a la piel pueden tener regresión espontánea después de algunos años.
Ya la esclerosis sistémica es una enfermedad de evolución lenta, pero permanente. El tratamiento tiene como objetivo aliviar los síntomas y reducir la actividad de la enfermedad.
Los fármacos utilizados dependen de los síntomas que el paciente tiene, por ejemplo:
- Vasodilatadores, como la nifedipina, se suele utilizar para tratar el fenómeno de Raynaud.
- Inhibidores de la bomba de protones como el omeprazol, ayudan en los síntomas gástricos y del reflujo.
- Antiinflamatorios y corticoides en dosis bajas ayudan en el dolor y en la rigidez de las articulaciones.
- Medicamentos inmunosupresores, como metotrexato o micofenolato mofetil, ayudan a reducir el daño de la piel y órganos causados por el sistema inmune.
- Antihipertensivos para tratar la hipertensión.
No hay tratamiento que sea universal para todos los tipos de esclerodermia. Cada tratamiento debe ser establecido de manera individual, según los síntomas y el tipo de compromiso del órgano existente en cada paciente.
Referencias
- 2013 classification criteria for systemic sclerosis: an American college of rheumatology / European league against rheumatism collaborative initiative – Annals of the Rheumatic Diseases.
- Update of EULAR recommendations for the treatment of systemic sclerosis – Annals of the Rheumatic Diseases.
- BSR and BHPR guideline for the treatment of systemic sclerosis – Rheumatology.
- Clinical manifestations and diagnosis of systemic sclerosis (scleroderma) in adults – UpToDate.
- Overview of the treatment and prognosis of systemic sclerosis (scleroderma) in adults – UpToDate.
- Pathogenesis of systemic sclerosis (scleroderma) – UpToDate.
- What is scleroderma? – Scleroderma Foundation.
- The case of frontal linear scleroderma (en coup de sabre) – Journal of Clinical and Analytical Medicine.



Dudas de los lectores sobre este tema
Preguntas reales enviadas por lectores y seleccionadas por el editor por su relevancia para este artículo.
Más comentarios de los lectores
Gracias por su ayuda. Mi diagnóstico es sclerosis sistemático progresivo. La información se relaciona a mi caso que desconocía. Desearía saber si encuentran la cura.