¿Qué es el retinoblastoma?
El retinoblastoma es un tumor maligno que se desarrolla en la retina, una fina capa compuesta de células nerviosas que recubre el interior del ojo y se encarga de captar la luz. La retina percibe la luz y envía impulsos a través del nervio óptico al cerebro, donde estas señales se interpretan como imágenes.
El retinoblastoma es el tumor ocular más común en niños y representa hasta el 15 % de todos los cánceres que aparecen en el primer año de vida. Aunque puede aparecer en ambos ojos, en más del 75 % de los casos el tumor se presenta en un solo ojo.
Este tumor ocular afecta a aproximadamente 1 de cada 15.000 niños. La edad promedio de diagnóstico es de 18 a 24 meses, siendo 12 meses para los niños con enfermedad bilateral y 24 meses para los niños con enfermedad unilateral. En el 95 % de los casos, el tumor aparece antes de los cinco años.
La incidencia es similar en niños y niñas, y no hay una predilección racial. Si no se trata, el retinoblastoma es una enfermedad mortal. Sin embargo, con los avances en el tratamiento, la supervivencia supera el 95 %.
El diagnóstico temprano y el tratamiento adecuado por un equipo multidisciplinario son esenciales para optimizar el resultado visual y la supervivencia del niño.
Origen del retinoblastoma
El retinoblastoma es una enfermedad de origen genético, que resulta de la mutación del gen RB1, ubicado en el brazo largo del cromosoma 13. Estas mutaciones provocan que las células de la retina continúen creciendo y multiplicándose de manera descontrolada, mientras que las células sanas mueren. Este exceso de células forma el tumor dentro del ojo.
Las células del retinoblastoma pueden invadir más profundamente y alcanzar estructuras alrededor del ojo. También pueden extenderse (metástasis) a otras áreas del cuerpo, como el cerebro y la columna vertebral.
Forma hereditaria
En el 30 % de los casos, la enfermedad es de origen hereditario. Esto significa que la mutación en el cromosoma 13 se hereda de uno de los progenitores. Si uno de los progenitores porta una mutación del gen RB1, cada hijo tiene un 50 % de probabilidades de heredar este gen.
Los niños con la forma hereditaria del retinoblastoma suelen desarrollar la enfermedad más temprano. A diferencia de la forma no hereditaria, el retinoblastoma hereditario a menudo afecta a ambos ojos.
Aunque la mutación del gen RB1 aumenta el riesgo de retinoblastoma, no garantiza que el niño desarrolle el cáncer.
Forma no hereditaria
El 70 % de los casos de retinoblastoma corresponde a la forma no hereditaria de la enfermedad, que siempre se manifiesta en un solo ojo.
Estos pacientes no son portadores de la mutación del gen RB1 en todas las células del cuerpo. Se cree que la mayoría de los niños con la forma no hereditaria del retinoblastoma desarrollan la enfermedad debido a daños en ambas copias del gen RB1 en una única célula retiniana en desarrollo.
Los pacientes que desarrollan la forma no hereditaria del retinoblastoma no corren el riesgo de transmitir el gen mutado a sus hijos.
Se recomienda el asesoramiento genético después del diagnóstico en un miembro de la familia para evaluar la necesidad de pruebas genéticas en otros miembros de la familia, especialmente hermanos, para determinar su riesgo de desarrollar la enfermedad.
Síntomas
El retinoblastoma a menudo se presenta con leucocoria, también conocida como «pupila blanca».
En los casos más evidentes, la leucocoria puede identificarse mediante una simple observación ocular, sin luz especial ni ninguna otra ayuda. Si se observa la pupila del niño, ésta presenta un color más claro.
En la mayoría de los casos, sin embargo, la pupila aparece blanca solamente en determinadas circunstancias, como cuando se dilata en una habitación poco iluminada, cuando el niño mira en una dirección específica o en fotografías con flash.
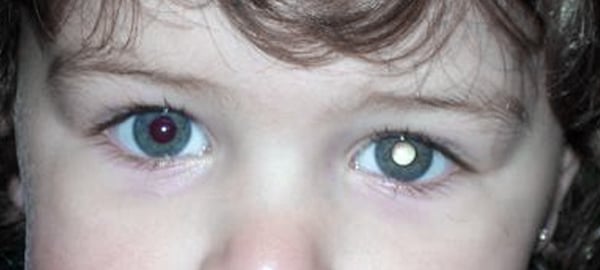
La leucocoria puede diagnosticarse al examinar los ojos con un oftalmoscopio. Sin embargo, la leucocoria no es exclusiva del retinoblastoma, puede ser causada por otras condiciones, como cataratas, desprendimiento de retina, retinopatía del prematuro, infección intraocular (endoftalmitis) y anomalías vasculares retinianas (enfermedad de Coats).
Además de la leucocoria, el niño puede presentar otros síntomas como estrabismo (ojos cruzados), nistagmo (movimientos oculares involuntarios), ojos rojos, dolor en los ojos, hinchazón ocular y disminución de la agudeza visual.
Historia natural del retinoblastoma
Si no se trata, el retinoblastoma puede ser fatal. El tumor crece dentro del ojo y puede destruirlo. La vía más común de propagación metastásica es a través del nervio óptico al sistema nervioso central. Las células tumorales también pueden invadir los vasos sanguíneos y diseminarse a los pulmones, los huesos, el hígado o el cerebro. Otra vía de propagación es a través del sistema linfático.
La diseminación metastásica suele ocurrir en los primeros 12 meses de la presentación clínica del retinoblastoma.
Con el tratamiento, la tasa de supervivencia del retinoblastoma supera el 95 %. Sin embargo, el pronóstico de la conservación del ojo es mucho más bajo y depende del estadio de la enfermedad en el momento del diagnóstico.
La regresión espontánea del retinoblastoma se ha documentado en un pequeño número de casos, pero es muy rara.
Estadios del retinoblastoma
El retinoblastoma se divide en dos estadios principales:
Retinoblastoma intraocular
La mayoría de los niños son diagnosticados en el estadio de enfermedad intraocular.
En este estadio, el cáncer se encuentra completamente dentro del ojo y no se ha extendido. Los tumores en este estadio se clasifican en los grupos A a E según el Sistema de Clasificación Internacional del Retinoblastoma Intraocular, en función de la gravedad de la enfermedad.
Grupo A:
- Riesgo muy bajo de pérdida del ojo u ojos afectados.
- Tumores pequeños, de 3 mm o menos.
- Los tumores se encuentran solamente en la retina, la capa de tejido ubicada en la parte posterior del ojo.
- Los tumores no están cerca de la fóvea (el «agujero» central de la retina) ni del nervio óptico.
- No hay tumores flotando en el ojo, lo que se conoce como siembra vítrea. La siembra vítrea ocurre cuando los tumores se extienden por el vítreo, un líquido transparente y gelatinoso que llena el ojo.
- No hay desprendimiento de retina, una situación de emergencia en la que la retina se desplaza de su posición normal.
Grupo B:
- Riesgo bajo de pérdida del ojo u ojos afectados.
- Uno o más tumores miden más de 3 mm.
- Los tumores solo están en la retina.
- No hay siembra en el vítreo.
- No hay desprendimiento de retina a más de 5 mm de la base del tumor.
Grupo C:
- Riesgo moderado de pérdida del ojo u ojos afectados.
- Los tumores están bien definidos.
- Los tumores se han extendido debajo de la retina, lo que se conoce como siembra subretiniana.
- Los tumores también pueden haberse extendido al vítreo.
- Hay desprendimiento de retina, con más de 5 mm desde la base del tumor hasta un desprendimiento de retina completo.
Grupo D:
- Existe un alto riesgo de pérdida del ojo u ojos afectados.
- La extensión del tumor es extensa hasta el vítreo/debajo de la retina.
- Lesiones vítreas o subretinianas de tipo «bola de nieve».
- Hay desprendimiento de retina, con más de 5 mm desde la base del tumor hasta un desprendimiento de retina completo.
Grupo E:
- Los retinoblastomas intraoculares clasificados en el grupo E han dañado extensamente el ojo.
- Las posibilidades de salvar el ojo afectado son casi nulas.
- Tumor(es) grande(s) que se extiende(n) hacia la parte anterior del ojo.
- Glaucoma neovascular. El glaucoma es un daño causado por la presión en el interior del ojo. En el glaucoma neovascular, esta presión se debe al crecimiento de nuevos vasos sanguíneos en la parte frontal del ojo como resultado del tumor.
- Hemorragia vítrea, es decir, sangrado del ojo.
- Encogimiento y atrofia del ojo.
- Hifema, que es la presencia de sangre en la parte anterior del ojo.
- Enrojecimiento e hinchazón intensos alrededor del ojo, similares a una infección, pero sin infección real.
Los retinoblastomas intraoculares de los grupos A a E son curables, pero el paciente puede perder el ojo si padece la enfermedad en los estadios más avanzados, especialmente D y E.
Retinoblastoma extraocular
El retinoblastoma extraocular es aquel que se ha extendido más allá del ojo, a los tejidos circundantes o a otras partes del cuerpo. Este estadio del tumor es poco frecuente en la actualidad, ya que la mayoría de los casos se diagnostican antes.
El retinoblastoma extraocular es mucho más difícil de curar y, si la enfermedad ya se ha extendido al sistema nervioso central, la tasa de supervivencia es inferior al 10 %.
Diagnóstico
Una vez que se sospecha de retinoblastoma, el niño debe ser remitido a un oftalmólogo para confirmar o descartar el diagnóstico. Algunas pruebas utilizadas con este propósito son:
- Mapeo retiniano o examen del fondo de ojo: en este examen, después de dilatar la pupila, el médico examina el ojo del niño con una lente especial tratando de identificar el tumor, pudiendo evaluar su tamaño y si hay afectación de otras estructuras del ojo.
- Ecografía ocular: esta exploración es esencial para evaluar el tamaño del tumor, si existe desprendimiento de retina y/o afectación de otras estructuras, como el nervio óptico, por ejemplo.
- Tomografía computarizada o resonancia magnética: ambas pruebas pueden ayudar a diagnosticar el tumor, pero son más importantes para evaluar la presencia de metástasis.
En niños pequeños, menores de 3 años, puede ser necesario realizar las pruebas con el niño sedado o bajo anestesia general.
Cribado
En los lactantes de alto riesgo, es decir, aquellos con antecedentes familiares positivos de retinoblastoma, la detección del tumor debe realizarse de manera rutinaria en los primeros años de vida.
La Asociación Americana de Oncólogos y Patólogos Oftalmólogos sugiere el siguiente esquema de cribado para el retinoblastoma:
- Durante los tres primeros años de vida, los exámenes de cribado se realizan inicialmente cada uno o dos meses y luego se espacian cada tres meses si no hay hallazgos preocupantes.
- Entre los tres y los siete años, las pruebas se realizan cada cuatro o seis meses.
- Si la prueba genética revela que el niño no tiene una mutación hereditaria del gen RB1, el cribado puede interrumpirse a los siete años.
- Si la prueba genética revela que el niño es portador de una mutación en el gen RB1, deberá seguir sometiéndose a pruebas periódicas cada uno o dos años a partir de los siete años.
Tratamiento
Como ya se ha mencionado, el retinoblastoma es una enfermedad curable. Por lo tanto, los objetivos del tratamiento son, en orden de prioridad: curar el tumor y salvar la vida del niño, conservar la visión y preservar el aspecto estético del ojo. No siempre pueden alcanzarse los tres objetivos.
Los mejores tratamientos para el retinoblastoma dependen del tamaño y la localización del tumor, de si el cáncer se ha extendido a otras zonas además del ojo, y del estado general de salud del niño.
Algunas opciones de tratamiento disponibles actualmente son la radioterapia, la braquiterapia, la quimioterapia, la crioterapia, la fotocoagulación con láser y la enucleación (extracción quirúrgica del globo ocular).
Los tumores de bajo riesgo (grupos A o B) suelen tratarse mediante fotocoagulación con láser o crioterapia. Los tumores de riesgo moderado (grupos C y D) se tratan con quimioterapia, braquiterapia o radioterapia y láser. En cambio, los tumores graves (algunos de los grupos D y E) solo suelen tratarse mediante la extirpación completa del globo ocular.
Cuando hay invasión del sistema nervioso central o metástasis a distancia, el tratamiento indicado es quimioterapia y radioterapia.
Dudas frecuentes
¿Hay cura para el retinoblastoma?
Sí, actualmente la tasa de curación del retinoblastoma es superior al 95 %.
¿Puede aparecer el retinoblastoma en adultos?
Puede, pero el retinoblastoma en personas mayores de 7 años es extremadamente raro.
¿Cuáles son los signos y síntomas del retinoblastoma a los que deben estar atentos los padres?
Los signos y síntomas típicos del retinoblastoma son:
• Una pupila que parece blanca, en lugar del negro normal. Este cambio es más evidente cuando una luz intensa, como el flash de una cámara, incide directamente sobre los ojos.
• Ojos desalineados o bizcos (estrabismo).
• Problemas de visión.
• Ojo rojo y doloroso.
• Pupila dilatada.
• Iris de distinto color (el iris es la parte coloreada del ojo).
¿Cuál es el gen relacionado con el retinoblastoma?
En los pacientes con retinoblastoma hereditario, el tumor se desarrolla como resultado de cambios en un gen específico conocido como RB1, que se localiza en el cromosoma 13 en la posición q14.1-q14.2.
El RB1 es el único gen asociado al retinoblastoma hereditario. La proteína producida por el gen RB1 actúa como «supresor tumoral», lo que significa que ayuda a evitar que las células crezcan y se dividan con demasiada rapidez. Si una persona hereda una alteración en el gen RB1, pierde esta protección y tiene más probabilidades de padecer retinoblastoma.
¿Quién que no tenga antecedentes familiares de retinoblastoma puede desarrollar la enfermedad?
Sí, es perfectamente posible y frecuente que un niño sea el primer caso de retinoblastoma en la familia.
¿Los pacientes con retinoblastoma hereditario tienen mayor riesgo de desarrollar otros cánceres?
Sí, los pacientes que heredan una mutación en el gen RB1 también tienen mayor riesgo de desarrollar sarcoma osteogénico, sarcomas de partes blandas (especialmente en las áreas que recibieron radioterapia) y melanoma. Aunque el riesgo de cáncer en estos pacientes es mayor que en la población general, la mayoría de los sobrevivientes de retinoblastoma no desarrollan un segundo cáncer.
- Retinoblastoma: Clinical presentation, evaluation, and diagnosis – UpToDate.
- Retinoblastoma: Treatment and outcome – UpToDate.
- Retinoblastoma – Health Professional Version – National Cancer Institute.
- What Is Retinoblastoma? – American Cancer Society.
- Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists – Ophthalmology.
- Retinoblastoma – Childhood: Stages – American Society of Clinical Oncology (ASCO).
- Yanoff M, et al., eds. Malignant intraocular neoplasms. In: Ophthalmology. 5th ed. Edinburgh, U.K.: Elsevier; 2019.
- Kliegman RM, et al. Retinoblastoma. In: Nelson Textbook of Pediatrics. 20th ed. Philadelphia, Pa.: Elsevier; 2016.







Dudas de los lectores sobre este tema
Preguntas reales enviadas por lectores y seleccionadas por el editor por su relevancia para este artículo.